Mª B. LÓPEZ ÁLVAREZ*, G. HAWKINS GONZALEZ**
Introducción
Se caracterizan por ser enfermedades neurodegenerativas con período de incubación muy largo, clínica de deterioro neurológico progresivo e irreversible y cambios anatomopatológicos característicos con pérdida neuronal, proliferación glial y aspecto espongiforme del cerebro (1).
Este tipo de enfermedades se han descrito tanto en animales como en humanos (Tabla 1).
El origen de estas enfermedades neurodegenerativas era atribuido hace unos años, a los denominados "virus lentos" y que hoy son identificadas como enfermedades producidas por priones.
Concepto de prión
Los priones y su patogenia fueron descritos por Stanley B. Prusiner, al referir la existencia de un agente infectivo de origen proteico y carente de ácidos nucleicos, que se denomina prión por ser una partícula proteica infectiva (2).
Existen proteínas priones celulares, PrPc, que son constituyentes normales de algunas membranas celulares como de las neuronas del sistema nervioso central (SNC), del sistema nervioso entérico y del tejido linfoide.
El prión patógeno, PrPSc, llamado así por derivar de proteína prión del scrapie, es una variante anómala de la proteína prión celular normal (3). La PrPc es una proteína de unos 250 aminoácidos con una estructura tridimensional donde predomina la configuración alfa. El gen PRNP, que expresa esta proteína está codificado en el brazo corto del cromosoma 20.
La PrPSc es una proteína con una configuración predominantemente beta. Parece ser que los priones infectivos actuarían modificando la estructura normal de la PrPc y esto pone en marcha un mecanismo de destrucción celular neuronal, proliferación de material proteico y reacción espongiforme posterior. Los priones patógenos se caracterizan por ser muy resistentes a los medios habituales de desinfección y a las proteasas (4).
Tabla 1.
Encefalopatías espongiformes
[b]En Humanos[/b]
Kuru.
Enfermedad de Creuztfeldt-Jacob: esporádica, hereditaria, yatrogénica, nueva variante.
Enfermedad de Gerstmann-Sträussler-Scheinker
Insomnio familiar fatal o mortal
[b]En animales[/b]
Scrapie, tembladera o modorra, en ovejas y cabras.
Encefalopatía Espongiforme Bovina o "mal de las vacas locas", en vacas
Encefalopatía Espongiforme felina, en gatos domésticos y salvajes.
Encefalopatía espongiforme de visones, en visones
Enfermedad crónica debilitante, en ciervos, alces y antílopes.
Encefalopatía de ungulados exóticos.
Encefalopatía espongiforme de animales de laboratorio.
Enfermedades producidas por los priones en animales y humanos
La primera descripción de una de estas enfermedades data del siglo XVIII, fue el Scrapie de las ovejas. A principios del siglo XX se da a conocer la Enfermedad de Creuztfeldt-Jacob. Es en la década de los cincuenta del siglo pasado cuando se describe otra de estas enfermedades, el Kuru señalándose el canibalismo como mecanismo de transmisión. En el año 1985 en Inglaterra se describe una nueva enfermedad que aparece en las vacas, es la Encefalopatía Espongiforme Bovina (EEB), y se observó
que en el estudio del cerebro de estos animales existían alteraciones similares a las enfermedades antes referidas y que se detectaban priones en tejido cerebral y que la inoculación de este material infectado a animales de laboratorio producía la enfermedad igual que sucedía en las otras encefalopatías espongiformes. En los años setenta se introduce el consumo de harinas de origen animal en
las explotaciones ganaderas, estas harinas animales fabricadas con restos de animales, entre otros ovejas enfermas de Scrapie fue el origen de esta enfermedad, posteriormente las vacas enfermas fueron tambien utilizadas para la fabricación de harinas animales y la epidemia se fue extendiendo por el Reino Unido y posteriormente a otros países europeos (5,6).
En el año 1996 se describe una enfermedad en humanos a la que se denomina nueva variante de Creutzfelt-Jacob (nvECJ) (7) y que se parece a la presentación clásica, si bien tiene algunas peculiaridades como aparecer en pacientes más jóvenes, predominio de clínica psiquiátrica y se relacionó con el consumo de carne de vacuno contaminada por EEB.
La mayoría de casos de nvECJ se han descrito en el Reino Unido, tres en Francia y uno en Irlanda. Se desconoce cuál puede ser la incidencia en el futuro de esta enfermedad.
En relación a la posible capacidad de contagio de los distintos tejidos de animales afectados, se han establecido distintas categorías, alta, media, baja y muy baja o no detectable (3), ver en la Tabla 2, modificada de la clasificación de la OMS. No está permitido el uso de sangre para transfusiones de personas sospechosas de contacto con material contaminado, ni se permite el uso de órganos para
trasplante de pacientes fallecidos por encefalitis.
Conviene reseñar que el diagnóstico exacto de las encefalitis espongiformes lo da la anatomía patológica y la demostración de la existencia de priones infectivos en tejidos cerebral y la capacidad de transmisión de la enfermedad a animales de experimentación tras la inoculación de tejido infectado. Kuru
Este término significa temblor en lengua Fore, y se habla en una zona de Nueva Guinea donde se describió una enfermedad endémica, llegó a afectar al 1% de la población, que se caracterizaba por ataxia cerebelosa severa, movimientos involuntarios asociados como coreoatetosis, mioclono y temblor y posteriormente deterioro mental progresivo y signos de liberación frontal. La muerte de la persona suele producirse al año del inicio de la clínica (11). El período de incubación es muy largo, hasta de 20-30 años.
La anatomía patológica del cerebro presenta: alteraciones espongiformes, placas de amiloide. La afectación predominante es de cortex cerebeloso, cerebral y núcleos grises de la base. Se estableció que el mecanismo de contagio era a través de ritos caníbales. La enfermedad era trasmisible a humanos, chimpancés y otras especies animales que ingerían restos cerebrales de afectados. Desde la supresión del canibalismo, no se han producido nuevos casos.
Tabla 2.
Grado de infectividad de órganos y tejidos
Alta Cerebro:, ojo, médula espinal
Media: Ileon, colon proximal, placenta, ganglios, amígdalas bazo, glándulas pituitaria y adrenal, líquido cefalorraquídeo
Baja: colon distal, médula ósea, hígado, pulmón, páncreas
Muy baja: sangre, saliva, leche, orina, piel, testículo, músculo esquelético, hueso, corazón, cartílago.
Enfermedad de Creutzfeldt-Jacob ( ECJ )
Enfermedad neurodegenerativa poco frecuente, su incidencia es de 1/1000.000. Su distribución es mundial si bien parece existir más incidencia en algunas regiones de Libia, Israel y Eslovenia.
Se han descrito tres formas (10,13): esporádica, es la más frecuente supone un 90% de los casos; familiar o hereditaria, es autosómica dominante, se calcula que es entre 5%-10% de los casos y la infecciosa, es rara y la transmisión se produce por material contaminado de enfermos, así sucede en trasplantes corneales, injertos de duramadre, a través de instrumentos de neurocirugía y electrodos
esteroatáxicos, tratamientos con hormonas procedentes de cadáveres (8). La enfermedad también es trasmisible a otras especies animales.
El tiempo de incubación es largo se calcula entre 18 meses a 30 años. La edad media de aparición suele ser entre los 57-62 años.
Clínica, la forma más frecuente de inicio es como una demencia presenil subaguda.
Los síntomas fundamentales de esta enfermedad son la demencia progresiva y las mioclonias, éstas suelen aparecer o empeorar si el paciente se sobresalta. Existen otros síntomas que varían de un paciente a otro en función de la región cerebral afectada, así puede haber signos piramidales, hiperreflexia, Babinski hasta en el 40%-80% de los pacientes. Signos extrapiramidales, hipocinesia, alteraciones cerebelosas como ataxia y/o nistagmus aparecen hasta en 60% de los pacientes. Son muy infrecuente las alteraciones del sistema nervioso autonómico, las alteraciones sensoriales, las crisis epilépticas y no hay alteración del sistema nervioso periférico.
Pruebas complementarias: no suelen existir anomalías en pruebas de hemograma y bioquímica, en algunos casos se han descrito alteraciones de transaminasas.
El análisis de líquido cefalorraquídeo suele ser inespecífico, si bien en la ECJ esporádica (12), suele detectarse la proteína 14-3-3, lo que ayuda al diagnóstico. Se han detectado aumentos de la proteína 14-3-3 en otra patologías como en encefalitis virales y metabólicas entre otras. Electroencefalograma (EEG), presenta un patrón característico, en algunos momentos de la enfermedad, con un enlentecimiento difuso de la actividad de fondo y complejos periódicos de ondas bifásicas o trifásicas a intervalos regulares, esto
aparece entre un 65%-95% de los pacientes. Resonancia Magnética Nuclear (RMN), se ha descrito en casi el 80% de los pacientes la existencia de un aumento de la señal T2 en los ganglios de la base y en el tálamo. Anatomía Patológica (AP), pérdida neuronal, gliosis reactiva, degeneración espongiforme y placas de amiloide en tejido cerebral.
El diagnóstico de la enfermedad lo darán la clínica, las pruebas complementarias señaladas y sobre todo la positividad de priones anómalos en tejido cerebral y la trasmisión de la enfermedad a animales de experimentación tras la inoculación del tejido cerebral infectado.
Nueva variante de la enfermedad Creutzfeldt-Jacob (nvECJ)
Se denomina así a una enfermedad de reciente aparición, el primer caso fue descrito en 1995 en Inglaterra y desde entonces han aparecido más de 70 casos en Inglaterra, tres casos en Francia y un caso en Irlanda. Esta enfermedad tiene gran interés porque se ha relacionado con la encefalitis espongiforme bovina, ya que su origen parece ser la ingesta de tejidos de animales que padecían la
enfermedad.
La clínica de la nvECJ (7) se diferencia de la de la ECJ clásica en distintos aspectos como:
-La edad de aparición es más temprana, media de edad de 29 años (16-48).
-El tiempo medio de supervivencia de la enfermedad es de unos 14 meses, más largo que la ECJ clásica (4-5 meses).
-La clínica debuta en más del 30% de los casos como un cuadro psiquiátrico, sea depresión, ansiedad, cuadros psicóticos, entre otros. Son frecuentes las alteraciones sensoriales en fase prodrómica. La ataxia, las alteraciones cognitivas, la inmovilidad y las mioclonias son más frecuentes en fases avanzadas de la enfermedad.
Pruebas complementarias (7), el análisis del LCR no es específico. Puede o no haber aumento de proteínas y/o células. No suele hallarse la proteína 14-3-3. La RMN puede ser normal o bien presentar un aumento de la intensidad de T2 en el tálamo. El EEG es anómalo en el 70% de pacientes al inicio de la enfermedad, pero suelen ser pequeños cambios no característicos.
La biopsia de tejido amigdalar (7) demuestra la existencia de priones (PrP), siendo este un método de diagnostico de la nvECJ. En la anatomía patológica del tejido cerebral, se aprecia la existencia de placas fibrilares, que se tiñen para PrPsc, formadas por un centro eosinofílico y alrededor cambios espongiformes. Esto se localiza sobre todo en cerebro y cerebelo (muy característico) y en ganglios
basales y tálamo. Todos los pacientes son homocigotos para la metionina en el codón 129.
Cada vez parecen hallarse más pruebas que apoyan la posibilidad de que la nvECJ no es sino la transmisión de la EEB desde las vacas a los seres humanos. El origen de la epidemia en animales en Gran Bretaña, la aparición de la mayoría de casos en la actualidad de nvECJ en este país.
La similitud entre las características de los priones en ambas enfermedades.
Se desconoce cuál será el grado de afectación a la población general que han consumido tejidos de animales enfermos pero muchos factores pueden influir como la barrera interespecie, la homocigosis para la metionina en el codón 129, el grado de infectividad de los distintos tejidos ingeridos (14).
Síndrome de Gertsmann-Straussler-Scheinker (SGSS)
Es una enfermedad con una incidencia muy baja 1-10 casos por 100 millones.
Es hereditaria, con patrón autosómico dominante (2). La edad media de aparición es de 45 años, y el tiempo medio de supervivencia es de unos 5 años (9,1).
La clínica se caracteriza por deterioro cerebeloso progresivo, asociado a distintos grados de demencia. Las diferentes formas clínicas de expresión de la enfermedad, predominantemente atáxica; ataxia-parkinson-demencia; cuadro pseudobulbar y demencia, etc, están en
relación con las alteraciones de aminoácidos en distintos codones del gen PRNP y del polimorfismo en el codon 129.
El EEG puede estar enlentecido o ser normal, no existen cambios específicos. El análisis del LCR no
muestra alteraciones. La RMN es inespecífica o puede haber un descenso de señal en T2 en el núcleo estriado y en mesencéfalo. En el estudio anatomopatológico del tejido cerebral se han hallado placas de tejido amiloideo sobre todo en cerebelo, pérdida neuronal en todo el cerebro y grado variable de espongiosis.
Insomnio familiar fatal o mortal (IFF)
Enfermedad muy rara (10). Es hereditaria, autosómica dominante. Todos los pacientes en el gen PRNP son homozigotos para la metionina en el codón 129 y tienen una mutación en el codón 178. La edad de presentación es de 35-61 años, la supervivencia es de 7-25 meses.
Lo más característico de la clínica (10,13) es el insomnio progresivo e intratable; signos disautonómicos, hiperhidrosis, hipertermia, taquicardia, hipertensión arterial, alteraciones endocrinológicas en la secreción de ACTH, cortisol, hormona del crecimiento, prolactina y melatonina. Puede haber otros síntomas y signos como: ataxia, mioclonias, alteraciones del lenguaje y de la memoria, confusión, alucinaciones o ilusiones. Es excepcional la demencia.
El estudio anatomopatológico del tejido cerebral muestra pérdida neuronal y astrogliosis en distintas regiones cerebrales.
Tratamiento de las enfermedades producidas por priones
No existe en la actualidad ningún tratamiento curativo, se han usado distintos fármacos para tratar a los pacientes, entre ellos Amantidina, Vidarabina, Aciclovir, Interferón, Anfotericina B, etc.., con escasos resultados.
En la actualidad se investiga sobre las características de los priones su estructura, patogenia, etc., y se ensaya con péptidos capaces de inhibir la conversión de los priones de formas normales a patológicas (15).
Bibliografía
1. Haywood AM. Transmisible spongiform encephalopathies. N Engl J Med 1997; 337: 1821-1828.
2. Prusiner SB. Biology and genetics of prion diseases. Annu Rev Microbiol 1994; 48:655-686.
3. Tan L, Williams MA, Khan MK, Champion HC, Nielsen NH. Risk of transmission of bovine spongiform encephalopathy to humans in the United States. JAMA 1999;281:2330-2339.
4. Gasset M, Westaway D. Los priones y su biología. Rev Neurol 2000; 31(2):129-132.
5. Prusiner SB. Prion diseases and the BSE crisis. Science 1997; 278:245-257.
6. Polo JM. Historia y clasificación de las enfermedades priónicas humanas. Rev Neurol 2000;31: 137-141.
7. Will RG, Zeidler M, Stewart GE, Macleod MA, Ironside JW, Cousens SN, Mackenzie J, Estibeiro K, Green AJE, Knight. Diagnosis of New Variant Creutzfeldt-Jacob Disease. Ann Neurol 2000; 47:575-582.
8. Milton H. How is Creutzfeldt-Jacob Disease Acquired?. Neuroepidemiology 2000; 19:55-61.
9. Budka H, Aguzzi A, Brown P, et al. Neuropathological diagnostic criteria for Creutzfeldt-Jacob disease(CDJ) and other human spongiform encephalopathies (prion diseases). Brain Pathol 1995;5:459-466.
10. Johnson RT, Gibbs J Jr. Creutzfeldt-jakob disease and related transmisible spongiform encephalopathies. N Engl J Med 1998; 339:1994-2004.
11. Gajdusek DC, Zigas V. Degenerative disease of the central nervous system in New Guinea: The endemic occurrence of "kuru" in the native population. N Engl J Med 1957;257:974-978.
12. Hsich G, Kenney K, Gibbs CJ, et al. The 14-3-3 brain protein
in cerebroespinal fluid as a marker for transmissible spongiform encephapathies. N Engl J Med 1996; 335:924-930.
13. Hernández-Albújar S, García-Tobaruela A, Torres Rodrígez E, et al. Priones: concepto y enfermedades. An Med Interna 1999;16:647-653.
14. Tyler KL. Risck of human exposure to bovine spongiform encephalopathy. BMJ 1995; 311:1420-1421.
15. Soto C, Kascsak RJ, Saborio GP et al. Reversion of prion protein conformacional changes by syntetic beta-sheet breaker peptides. Lancet 2000; 355:192-197.
viernes, 22 de abril de 2011
El ser humano: un verdadero superorganismo
LONDRES.- El ser humano promedio es más microbio que mamífero, un verdadero superorganismo que comprende 10 veces más células microbianas que humanas. Se piensa que el número total de genes microbianos en nuestro cuerpo excede el de genes humanos en una proporción de 1000 a 1. Con tantos parásitos uno puede afrontar el desparramar unos cuantos, pero estaría en problemas si no tuviera ninguno. De hecho, uno no sería humano, una paradoja que los científicos tratan de dilucidar. El único momento en que estamos libres de ellos es durante los 9 meses que pasamos dentro del vientre materno. Luego, somos lanzados al mundo de los gérmenes. | |
Pero una vez que los más fuertes han reclamado su lugar, las condiciones favorables que propician abren la puerta a sucesivas oleadas de migrantes, provenientes de otras personas, de los animales, de la casa; en realidad, de todo lo que el bebe toca y no es estéril. Dentro de los primeros años de vida, los niños adquieren una colonia microbiana estable en todas las partes del cuerpo, excepto en el cerebro, los riñones, la sangre y los pulmones. Luego, microbios y anfitrión coexisten por el resto de sus vidas. | |
Sin embargo, nuestras colonias estables no son meros pasajeros. Dependemos de ellas para sobrevivir. Los microbios normales -que incluyen virus, hongos, protozoos y bacterias- nos brindan un escudo contra gérmenes más desagradables, que están en el ambiente. Y, más aún, sin nuestros microbios normales no podríamos realizar muchas funciones corporales. Sin embargo, si se perturba la armonía natural, nuestros microbios aparentemente benignos pueden enfermarnos. Por eso los científicos quieren saber exactamente qué hay allí y cómo nuestros huéspedes microbianos interactúan entre ellos y con nosotros. |
Hasta hace poco, nuestro conocimiento era limitado, en parte porque sólo era posible identificar los microbios que podían ser cultivados en laboratorio, un mero 1 o 2 por ciento del total. Sin embargo, en años recientes se ha visto una explosión de conocimiento acerca de los habitantes microbianos de algunas partes de nuestro cuerpo, posible gracias a las nuevas herramientas de la biología molecular. Los microbiólogos pueden analizar hoy los genomas de comunidades microbianas enteras al cortar sus ADN en pequeñas partículas y realizar secuencias de esos fragmentos, para luego conectar las piezas como si reordenaran un rompecabezas. |
Hoy, esta área, conocida como metagenómica, está lista para despegar. Los Institutos Nacionales de Salud, de los Estados Unidos, aprobaron un plan de cinco años para investigar el microbioma humano, que es el contenido total microbiano del cuerpo humano. |
Estos microbios han evolucionado dentro de nosotros a través de milenios. En los bordes, sin embargo, hay un flujo que cambia constantemente en respuesta a las condiciones ambientales. Como resultado de ello, los científicos están arribando a la idea de que hay una continuidad entre nuestros microbios internos y los que habitan en el mundo exterior. Con este cambio de visión se llega a la conclusión de que podría ser posible manipular el microbioma humano para mejorar nuestra salud. El yogur probiótico que contiene bacterias vivas es sólo el comienzo; hay proyectos que incluyen a las bacterias que producen caries y microbios que ayudan a perder peso. |
Otra idea es que la huella microbiana de cada persona provee un registro histórico que puede ser utilizado para señalar el origen de nuestros antiguos ancestros. Y cómo la composición total del microbioma humano rastrea nuestra evolución conjunta con sus microbios refleja también los cambios que hemos tenido en nuestra manera de vivir. Desde el comienzo del siglo XX, y más notablemente desde la introducción de los antibióticos, nuestro microbioma ha estado cambiando más rápido que nunca. Si esto finalmente es bueno o malo para la salud humana, lo dirá sólo una mejor comprensión del microbioma humano. |
El zoológico cutáneo Uno de los objetos de estudio es la piel, que comprende seis o siete hábitats diferentes, desde los espacios húmedos entre los dedos del pie hasta la parte externa de la oreja. "No hay que asombrarse de que sea un verdadero zoológico", asegura Martin Blaser, de la Universidad de Nueva York. { En febrero, Blaser publicó el informe más completo sobre la piel que jamás se haya realizado. |
Aun así, sólo observó una pequeña superficie del antebrazo, donde encontró 240 especies en seis voluntarios sanos mediante una técnica que identifica diferentes microbios sobre la base de comparaciones genéticas. Al incrementar su muestra a 12 personas, Blaser contabilizó 360 especies. También descubrieron que no hay dos individuos con el mismo componente microbiano y que éste hasta cambia con el paso del tiempo en cada persona, pero todos comparten un núcleo o andamio bacteriano. Dado que la piel es el límite entre uno y el mundo exterior, es poco sorprendente que su comunidad microbiana sea tan variable. El equilibrio es extremadamente sensible a fluctuaciones ambientales y cambia cada vez que uno toma una ducha o hasta cuando se usa una nueva marca de jabón. |
A pesar de que una gran variedad de microbios puede vivir en la piel sin causar ningún daño, no siempre son benignos. Estas bacterias pueden producir infecciones si penetran en la sangre, dice Mike Wilson, microbiólogo del University College de Londres. Pero ninguno es tan frecuente como el Acné propionibacterium , una bacteria anaeróbica que se alberga en las glándulas y poros pobres en oxígeno y que causa el acné. Este microbio también ofrece una saludable lección a cualquiera que desee manipular la colonia microbiana de la piel. LA NACIÓN-19 DE AGOSTO 2007 |
domingo, 17 de abril de 2011
Lo que hay que saber del dengue
Dengue.
El virus pertenece a la familia Flavovirideae, junto con otros virus de importancia actual en patologías humanas como por ejemplo, el de la fiebre amarilla. En la transmisión intervienen mosquitos o garrapatas que pueden mantener al virus en la naturaleza, además de los reservorios animales que convierten la mayoría de las enfermedades por flavovirus en zoonosis (enfermedad transmitida por animales vertebrados e invertebrados y el hombre).
Una de las excepciones es el dengue, ya que el hombre es el único reservorio conocido.
Existen cuatro serotipos de virus del dengue:
- Tipo I (DEN 1)
- Tipo II (DEN 2)
- Tipo III (DEN 3)
- Tipo 4 (DEN 4)
La infección por alguno de los serotipos no produce inmunidad protectora cruzada, por lo cual la reinfección por otro serotipo es posible y además condicionaría la aparición del dengue hemorrágico.
Algunos subtipos del serotipo DEN 3 se han asociado con mayor frecuencia al dengue hemorrágico.
Es decir, si una persona es picada por el mosquito y contrae dengue, por ejemplo serotipo 1 y se vuelve a re infectar con serotipo 1, 2 o 3, es muy probable que en esta segunda infección desarrolle dengue hemorrágico en casi un 80 0 90% de los casos.
Es transmitido por la picadura de la hembra (hematófaga), introducido en América desde África por el comercio marítimo, en especial esclavos, adaptado al domicilio humano se lo encuentra en contacto íntimo con el hombre.
La hembra una vez fecundada, deposita los huevos en pequeñas colecciones de agua, en condiciones apropiadas de humedad y temperatura, en 48 horas eclosionan larvas que en 7 días maduran, pudiendo aparearse y repetir el ciclo. En condiciones no aptas los huevos depositados pueden permanecer viables durante un año.
La ovipostura causa un incremento metabólico en la hembra que la induce a la alimentación de sangre.
La picadura a un enfermo con dengue en período virémico infecta al mosquito que a su vez puede transmitir la infección al otro hospedador.
La magnitud de la epidemia del dengue en el mundo está estrechamente relacionada con las condiciones que favorecen la procreación y supervivencia de Aedes aegypti.
En América, desde el sur de EEUU y hasta la región pampeana, todos los países, excepto Chile y Canadá, tienen altos índices de infección por Aedes aegypti.
La OMS estima que 80 millones de personas se infectan por año en el mundo, siendo las zonas de mayor prevalencia el sudeste asiático, Pacífico Occidental, islas del Pacífico Sur, América Central y Sur.
Manifestaciones clínicas: la infección por el virus del dengue puede dar lugar a las siguientes eventualidades clínicas:
Ø Infección sin enfermedad manifiesta: sólo detectable por el estudio de la respuesta inmune, una gran cantidad de infectados son asintomáticos en el curso de una epidemia.
Ø Síndrome febril indiferenciado: cursa con fiebre y escasa repercusión general, de corta duración y sin complicaciones.
Ø Dengue clásico: el período de incubación es de 6 a 8 días, el comienzo es brusco, hipertermia que a veces responde poco o nada a los antitérmicos. Cefalea intensa, fotofobia y mialgias en miembros. Artralgias frecuentes. Éste síndrome gripal carece de afectación de las vías aéreas, dato importante que lo diferencia de la infección por virus de la influenza y parainfluenza. La convalecencia es prolongada caracterizada por astenia, cefalea y mialgias residuales que puede durar meses.
Ø Dengue hemorrágico y dengue con shock: se asume que estos pacientes han padecido (6 meses o antes) una infección por un serotipo de virus distinto. Los primeros días de la enfermedad son indistinguibles del dengue clásico, con el descenso de la fiebre el paciente refiere dolor abdominal intenso, náuseas y vómitos hasta el comienzo de las hemorragias. La profundización de los síntomas circulatorios y neurológicos indican estado de shock profundo. Las complicaciones se relacionan con la sobreinfección bacteriana del aparato respiratorio sepsis. Las causas de muerte son el shock, hemorragias masivas y las sobreinfecciones
Diagnóstico: La sospecha se basa en la epidemiología y las manifestaciones clínicas, el diagnóstico virológico consiste en el asilamiento del virus del suero o de muestras de tejidos de autopsia.
La profilaxis incluye el control de Aedes aegypti, saneamiento ambiental, el control químico es secundario a las acciones de saneamiento ambiental.
Algunos datos epidemiológicos.
Provincia de Santa Fe…
Hasta la fecha, se confirmó circulación viral de dengue en la localidad de Romang, provincia de Santa Fe y no se registran otras zonas con circulación viral de dengue en Argentina.
El serotipo hallado en Romang fue DEN 1 y hasta la fecha fueron notificados al Sistema Nacional de Vigilancia 98 casos (total provincial al 31/03/2011).
En Rosario…
Las autoridades sanitarias de nuestra ciudad informaron la confirmación de dos casos de dengue contraídos uno, por un hombre proveniente de la zona de la triple frontera, y el otro, una mujer que recientemente viajó a Paraguay.
En las áreas donde se registraron casos de dengue se están estudiando otros pacientes febriles detectados a partir de la búsqueda activa, y se continúa con el descacharrado y limpieza de calles; la búsqueda de larvas y tratamiento focal de criaderos; y los bloqueos de foco y rociado espacial en la zona donde residen los enfermos, acciones que permiten limitar el impacto de la circulación del virus, la cantidad de enfermos y circunscribir las áreas afectadas, no habiéndose propagado hasta el momento a nuevas zonas.
En Argentina…
El Ministerio de Salud de la Nación reconoció oficialmente que en el país ya hay 1460 casos estudiados de los cuales 129 fueron positivos para dengue, distribuidos según de muestra en la tabla 1.
PROVINCIA | Dengue CONFIRMADO | Total estudiados | ||
BUENOS AIRES | 9 | 69 | ||
CAPITAL FEDERAL | 4 | 33 | ||
CATAMARCA | 13 | |||
CHACO | 1 | 117 | ||
CORDOBA | 28 | |||
CORRIENTES | 48 | |||
ENTRE RIOS | 28 | |||
FORMOSA | 53 | |||
JUJUY | 194 | |||
LA RIOJA | 4 | |||
MENDOZA | 1 | 2 | ||
MISIONES | 192 | |||
NEUQUEN | 1 | 3 | ||
SALTA | 15 | 240 | ||
SAN JUAN | 1 | |||
SAN LUIS | 3 | |||
SANTA FE | 98 | 400 | ||
SGO. DEL ESTERO | 4 | |||
TUCUMAN | 28 | |||
TOTAL | 129 | 1460 |
Tabla 1: Notificaciones acumuladas de pacientes estudiados para dengue confirmados y estudiados totales. Argentina: 01/01/2011 al 31/3/2011.
n = 1460
Fuente: Ministerio de Salud. Boletín de vigilancia epidemiológica N° 72 – Argentina (01/04/2011
Algunos datos alarmantes…
El intendente de la ciudad de Charata, provincia de Chaco, confirmo la muerte de dos mujeres que contrajeron la enfermedad en esa ciudad que cuenta con 30 mil habitantes y en donde se estima extraoficialmente que más de 11 mil habitantes estarían infectados… (La Nación, 31/03/2011)
Notificación oficial de países limítrofes (casos de dengue por país – año 2011)
PAIS | Casos de dengue | Confirm. laborat. | Serotipos |
Bolivia | 7842 | 1587 | DEN 1-2-3 |
Brasil | 155613 | 0 | DEN 1-2-3-4 |
Paraguay | 13175 | 2495 | DEN 1-2 |
Fuente: Ministerio de Salud. Boletín de vigilancia epidemiológica N° 72 – Argentina (01/04/2011)

Mapa epidemiológico para denge a nivel mundial
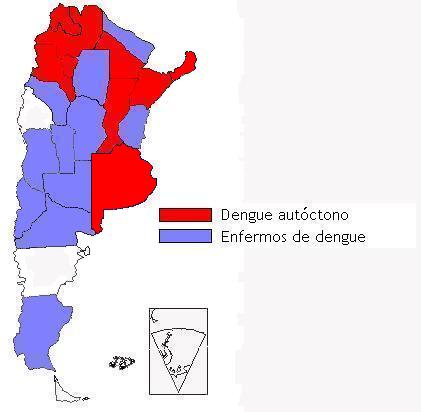
Mapa epidemiológico para dengue en Argentina
Conclusión...

Suscribirse a:
Entradas (Atom)